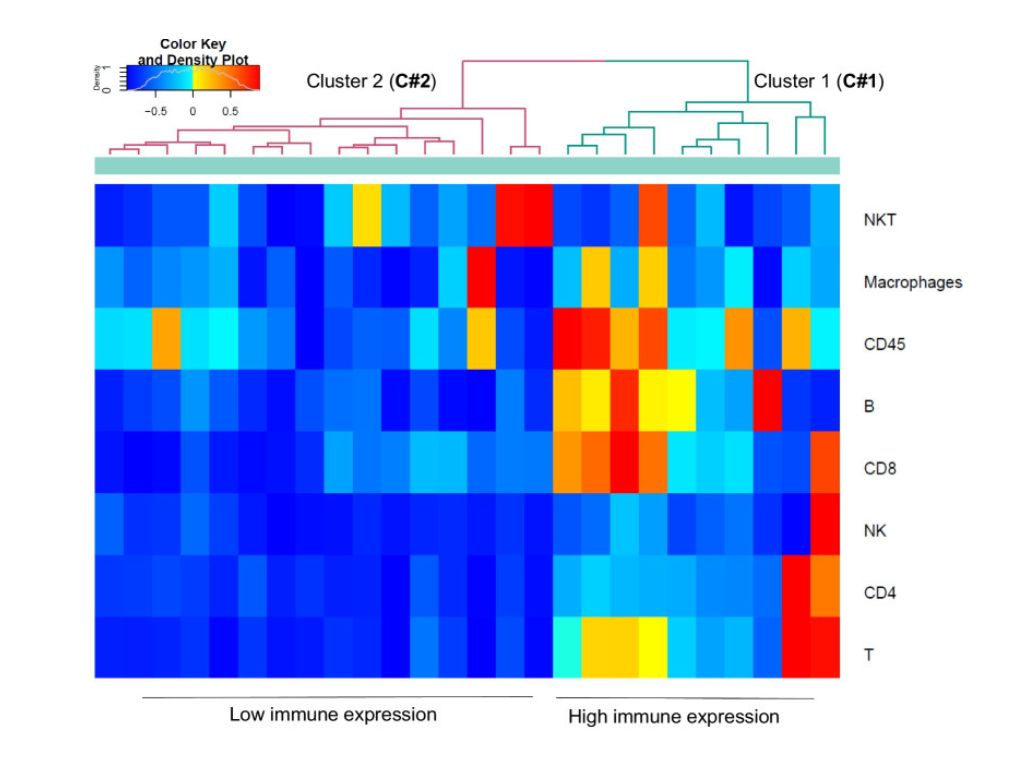
Cellular Senescence Markers and Matrix Metalloproteinase-7 Are Co-expressed in Fibrosing Interstitial Lung Diseases
Study highlights the role of cellular senescence markers (p16, p21) and MMP-7 co-expression in fibrosing interstitial lung diseases.
Impact of Ex Vivo Lung Perfusion System on the Lung Microbiome
Discover how ex vivo lung perfusion (EVLP) impacts the lung microbiome and its implications for improving donor lung viability and reducing transplant complications.
Falling through the cracks: what happens to survivors of preterm birth?
Survivors of preterm birth face increased risks of respiratory diseases, yet awareness among specialists is low. This study examines gaps in long-term care, highlighting the need for clear follow-up guidelines and improved communication between medical teams.